COMPARATIVE ANALYSIS OF Y449 MUTANTS
This mutation site was located in the core region of the binding interface, and corresponding mutations were more likely to experience structural perturbations in protein structure. Amino acid Y449 of 2019-nCoV-S1 formed good hydrogen bonds with neighboring Q42 and D38 on ACE2 (Figure 2A). Thus the structural stability of mutants would be significantly affected by residues Q42 and D38:
(a) Hydrophobic residues CGPAVILMF (Figure 2B/C), non-charged polar amino acids STNQ and negatively charged residues DE (Figure 2D) all could not compensate for the polarities of the hydrogen bonds between Y449 and Q42/D38. As a result, with the disappearance of wild-type polar interactions, the conformation of each mutant and corresponding RMSD value had experienced obviously variation.
(b) The positive charged amino acids HKR were not able to form hydrogen bonds between Y449-Q42/D38, but could form new salt-bridge interactions with residue D38 on ACE2 protein. In particular, mutated residues K and R could induce the position deflection of Q42, and new polar contacts network (H-bonds and salt-bridges) were formed (Figure 2E/F/G). These 3 mutants (Y449R/K/H) were selected for subsequent analysis.
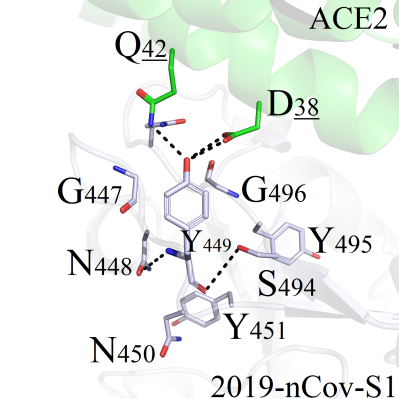 A
A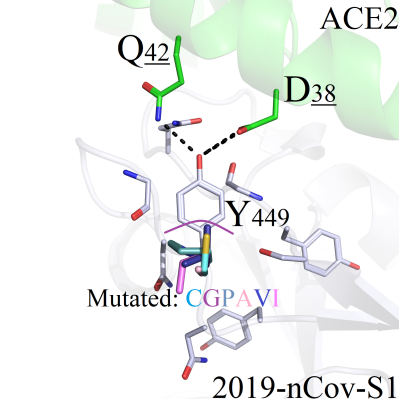 B
B C
C D
D E
E F
F G
GFigure 2. Binding pattern comparisons of Y449 series mutants. (A) Interaction modes between Y449 and surrounding amino acids; (B) Binding modes comparsions between Y449 mutants (Y449C/G/P/A/V/I) and wild-type protein; (C) Binding interactions of Y449 mutant series (Y449L/M/F) and wild-type complex; (D) Binding interactions of the wild-type and Y449 mutants (Y449D/E/N/Q/S/T); (E) Interaction pattern of the wild-type and mutant Y449H; (F) Interaction pattern of the wild-type and mutant Y449K; (G) Binding modes of the wild-type and mutant Y449R.