| • BiXGBoost | |
|---|---|
|
| • DyNetViewer | |
|---|---|
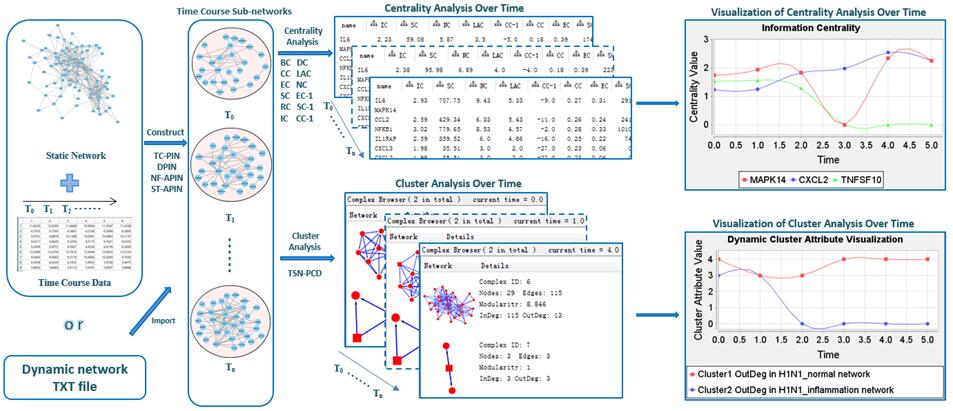
|
| • CytoCtrlAnalyser | |
|---|---|
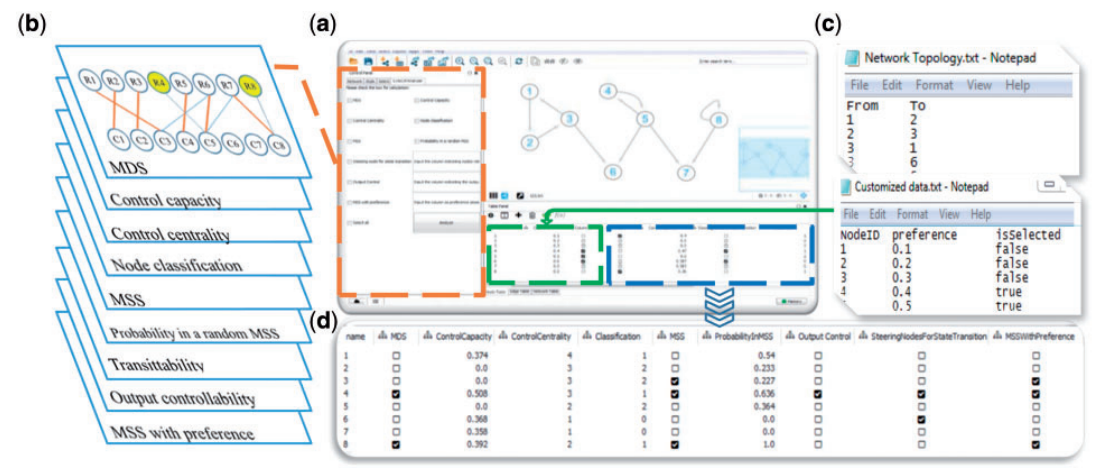
|
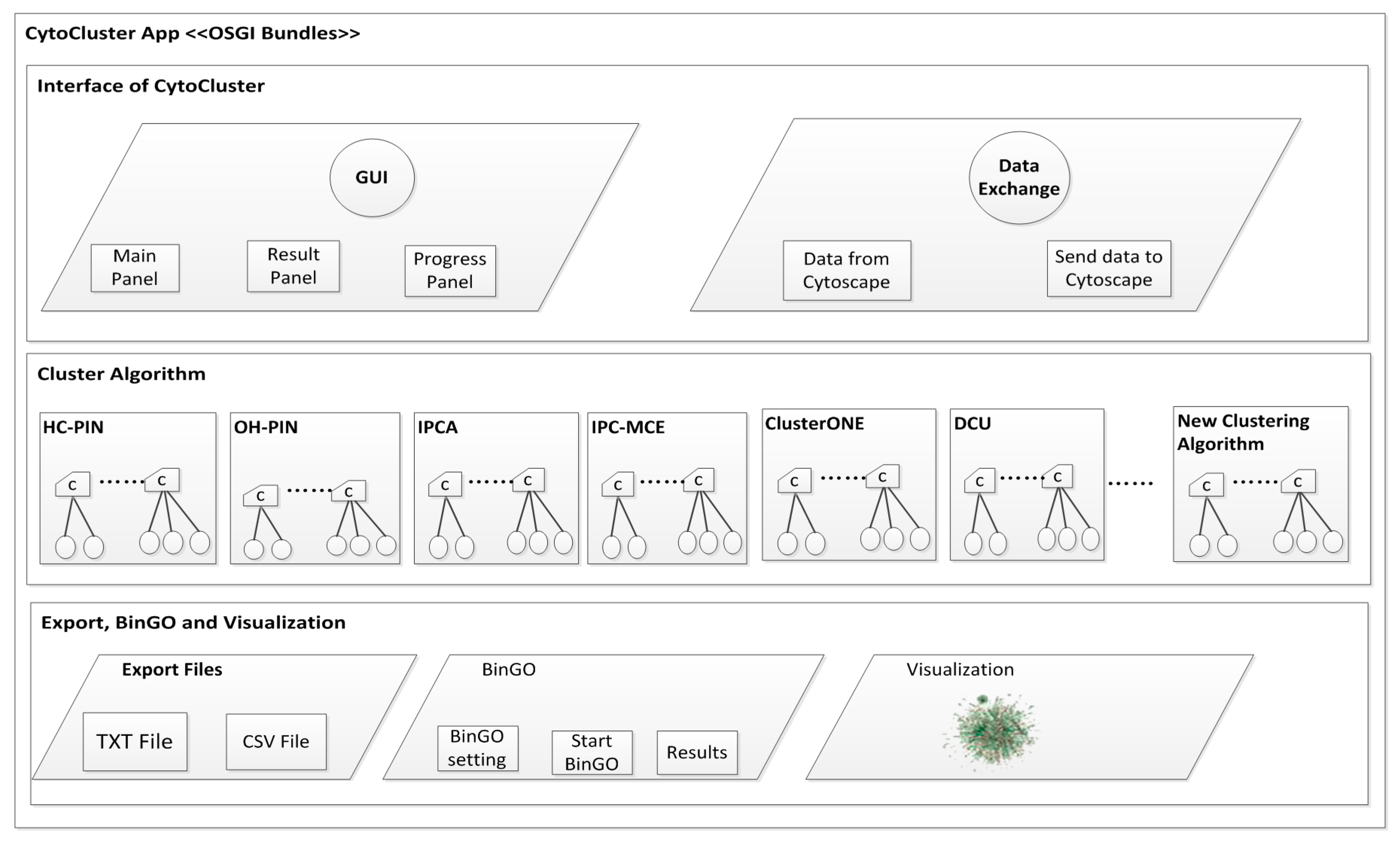
| • Cytocluster | |
|---|---|
|
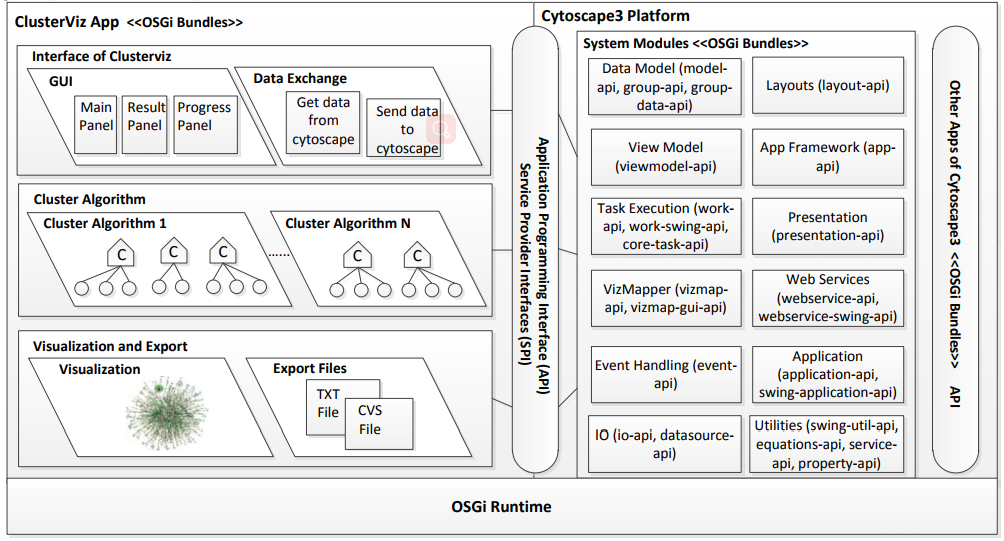
| • ClusterViz | |
|---|---|
|
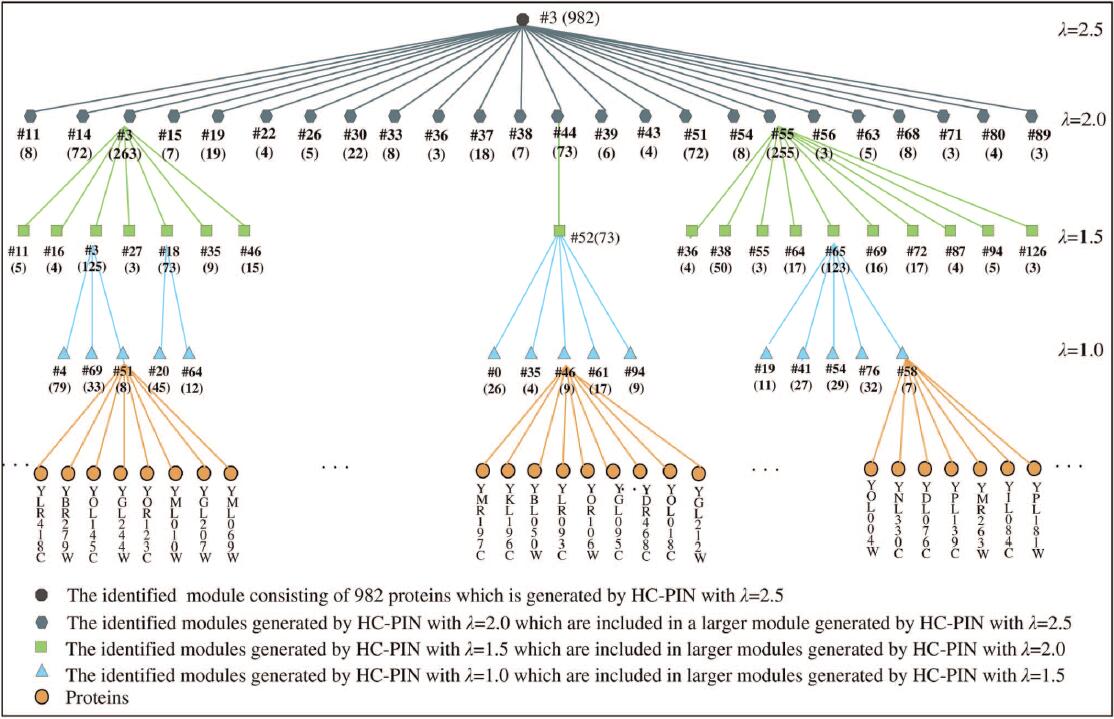
| • HC-PIN | |
|---|---|
|
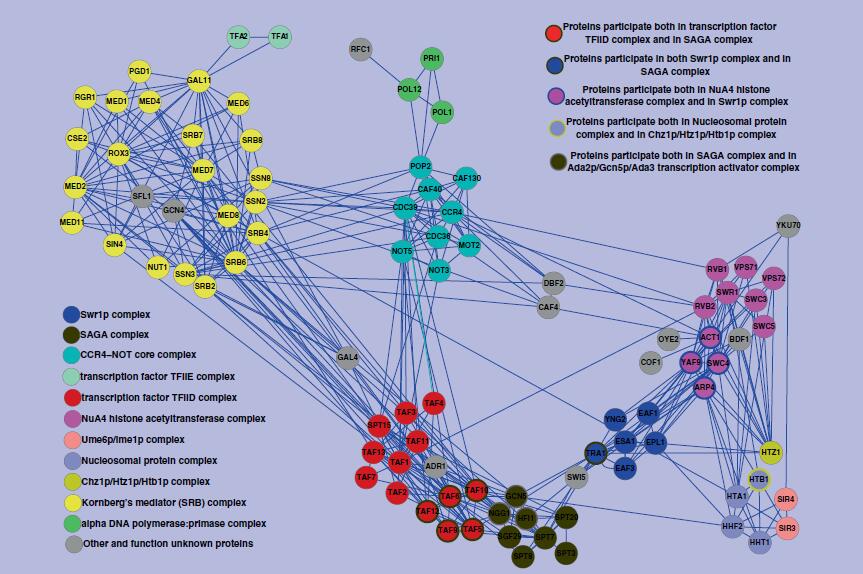
| • DFM-CIN | |
|---|---|
|
DFM-CIN is a new framework to distinguish between protein complexes and functional modules by integrating gene expression data into protein-protein interaction (PPI) data. A series of time-sequenced subnetworks (TSNs) is constructed according to the time
that the interactions were activated.
[Link]
[Paper]
|
| • IPCA |
|---|
| • CytoNCA | |
|---|---|
|
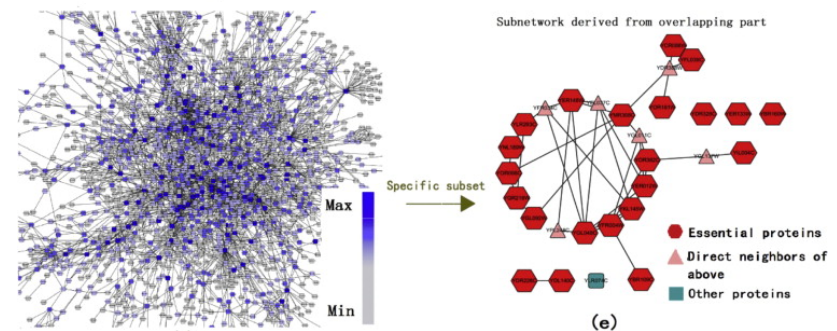
CytoNCA supports eight different centrality measures and each can be applied to both weighted and unweighted biological networks.It allows users to upload biological information of both nodes and edges in the network, to integrate biological data with
topological data to detect specific nodes.
[Link]
[Paper]
|
| • CPPK&CEPPK | |
|---|---|
|
| • PeC |
|---|
| • LAC |
|---|
| • PROBselect | |
|---|---|
|
| • DeepDISOBind | |
|---|---|
|
| • DeepPFP-CO |
|---|
|
DeepPFP-CO uses Graph Convolutional Network to explore and capture the co-occurrence of GO terms to improve the prediction of protein function.
[Link]
|